
Arino
Los arinos son intermedios de reacción neutros extremadamente reactivos. Pueden considerarse como derivados de sistemas aromáticos por la pérdida formal de dos sustituyentes.[1][2] La manera más habitual de representar un arino es mediante un triple enlace formal tensionado, sin embargo también es conocido su carácter dirradical o su estructura cumulénica. Aunque el término arino se asocia comúnmente al orto-arino, 1,2-dideshidroareno, los dideshidroderivados 1,3- y 1,4- también son conocidos.
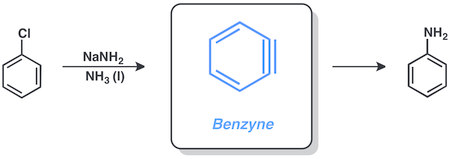
El descubrimiento de los arinos condujo a un rápido desarrollo de distintas metodologías sintéticas para sacar provecho de estos intermedios en síntesis orgánica. Una gran variedad de productos naturales han sido preparados mediante el uso de la química de arinos.[3]
Enlace[editar]
La estructura más utilizada para respresentar los arinos consiste en un triple enlace formal tensionado, sin embargo sus formas resonantes dirradical o cumulénica, también han sido descritas.[4]

Las restricciones geométricas sobre el triple enlace del orto-bencino conllevan la disminución de la superposición de los orbitales p en el plano, debilitando el triple enlace formal.[5] Rasziszhewski propuso una frecuencia de vibración del triple enlace del bencino en1846 cm-1,[6] siendo mucho más débil que la de un triple enlace en un alquino sin restricción cuya frecuencia vibratoria sería aproximadamente de 2150 cm-1. Con todo, el alquino tensionado es la representación más adecuada de la naturaleza del triple enlace del orto-bencino, más que la dirradical (mayor gap entre los estados singlete y triplete y reactividad de tipo alquino).[7] Las restricciones geométricas suponen igualmente una reducción significativa de la energía del LUMO de los arinos (desde 6.41 eV para el 2-butino hasta 1.33 eV en el bencino) mientras que la energía del HOMO permanece prácticamente constante según diversos estudios computacionales.[5][8]

El orbital LUMO de los arinos es de menor energía que el de los alquinos no tensionados, lo que implica una mayor cercanía al HOMO de nucleófilos. Se deduce por lo tanto que el bencino posee cierto carácter electrófilo y experimenta reacciones con nucleófilos.[9] Un análisis detallado de OM del bencino se presentó en 1968.[10]

Historia[editar]
El primer indicio de la existencia de los arinos como intermedios se debe al trabajo de Stoermer y Kahlert en1902. Observaron que al tratar con una base el 3-bromobenzofurano en etanol se formaba el 2-etoxibenzofurano. Sobre la base de este resultado se postuló un intermedio llamado arino.[11]

Georg Wittig y colaboradores propusieron que la formación de bifenilos a través de reacciones de fluorobencenos con fenillitio se llevaba a cabo gracias a un intermedio zwitteriónico[12][13][14] que fue confirmado experimentalmente por John D. Roberts en 1953.[15][16][17][18][19]
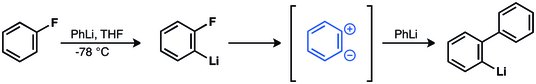
En 1953 John D. Roberts realizó un experimento clásico de marcado con 14C que reforzó la hipótesis de la existencia del bencino.[15] Roberts y sus estudiantes hicieron reaccionar clorobenceno-1-14C con amiduro de potasio con el fin de analizar la anilina resultante. Observaron que se producía igual cantidad de anilina con incorporación de 14C en los carbonos C-1 que en el C-2. Este resultado implica la formación de un intermedio simétrico, conocido ahora como bencino.

Poco después del hallazgo de Roberts, Wittig y Pohmer descubren que los bencinos pueden participar en reacciones de cicloadición [4+2].[20]

Otras evidencias de la existencia de esta especie son los estudios espectroscópicos: IR,[21] UV/Vis,[22] microondas[23] y espectros de RMN[24],.[25] También se ha observado en el interior de un “contenedor molecular”.[25][26]
Generación de arinos[editar]
Debido a su alta reactividad, los arinos deben generarse in situ. Los primeros métodos de obtención de arinos utilizaban condiciones severas como bases fuertes o altas temperaturas. Los métodos clásicos utilizan haluros de arilo tratados con una base fuerte para eliminar un protón aromático y generar el bencino a través de una eliminación.

El 2-carboxilato de bencenodiazonio puede servir como precursor de los bencinos. El principal inconveniente de este método es la naturaleza explosiva de las sales de diazonio.

Se fueron desarrollando métodos más suaves para la generación de arinos. En la última década, el método más empleado utiliza triflatos de arilo para la generación de bencinos. El ataque de los fluoruros al grupo trimetilsililo seguido de la eliminación del grupo triflato genera el bencino en condiciones suaves.

Las reacciones Deshidro-Diels-Alder (DDA) se describen como cicloadiciones de un 1,3-diino y un alquino. Estas reacciones generan un bencino que puede dar lugar a diversos derivados del benceno.[27]

Reactividad de los arinos[editar]
Incluso a bajas temperaturas los arinos son altamente reactivos. Esta reactividad se puede clasificar en 4 grandes grupos: (1) adiciones nucleófilas, (2) reacciones pericíclicas, (3) inserciones en enlace σ y (4) reacciones catalizadas por metales.
Adiciones nucleófilas[editar]
El tratamiento de haluros de arilo con bases fuertes produce una deshidrohalogenación que origina el arino correspondiente. El bencino así obtenido puede llevar a la formación de productos de adición nucleófila. La generación del intermedio arínico suele ser la etapa lenta en este tipo de reacciones.[28]

Las reacciones de acoplamiento de arinos permiten la generación de compuestos de tipo bifenilo. Algunos de estos compuestos son de gran importancia en la industria farmacéutica, en agricultura e incluso en la síntesis de ligandos para una gran variedad de transformaciones catalizadas por metales.[29]

Regioquímica de la formación del triple enlace formal[editar]
Cuando un grupo saliente (LG) y un sustituyente (Y) se encuentran en orto o en para, la generación del arino conduce a un único intermedio de este tipo. Sin embargo cuando el grupo saliente se encuentra en meta a otro sustituyente, existen dos posibilidades distintas dando lugar a los intermedios arínicos A y/o B. Si el arino (con un sustituyente en meta) se genera mediante el tratamiento con una base fuerte, será la acidez de los protones situados en orto al grupo saliente quien dirija la selectividad en generación del arino (no teniendo en cuenta factores estéricos). Por tanto, si Y es un grupo aceptor de electrones, entonces HB es más ácido que HA dando lugar al regioisómero B. De manera análoga, si Y es un grupo dador de electrones el regioisómero A será el generado, dado que HA es el protón más ácido.

Regioquímica de la adición nucleófila al triple enlace formal[editar]
Existen dos posibles regioisómeros de un arino bencénico con sustituyente Y: el triple enlace se puede situar entre los carbonos C2 y C3 o entre los carbonos C3 y C4. En caso de que el triple enlace se encuentre entre los carbonos C2 y C3, sustituyentes aceptores de carga electrónica (electron withdrawing substituents EWG) dirigirán la adición nucleófila para formar el carbanión más cercano al EWG. Sin embargo si se trata de sustituyentes dadores de carga electrónica (electron donating substituents EDG) proporcionarán poca selectividad entre los dos productos. Si el triple enlace se encuentra entre los carbonos C3 y C4, el efecto de los sustituyentes en la adición nucleófila se ve disminuido y se obtendrá frecuentemente una mezcla de productos para y meta.[28]

Ejemplos de síntesis total[editar]
Las adiciones nucleófilas a arinos han sido ampliamente utilizadas en la síntesis de productos naturales. En realidad es una de las aplicaciones más tradicional en este tipo de química. Se utilizaron, por ejemplo, en la síntesis de los siguientes productos: criptaustoline (1) y criptowoline (2).[30]

La síntesis del meroterpenoide tetracíclico (+)-liphagal conlleva la formación de un intermedio de tipo arínico. La aproximación sintética implica una ciclación con un arino para cerrar el anillo heterocíclico del producto natural.[31][3]

Las reacciones multicomponentes de arinos son poderosas transformaciones que permiten una rápida formación de arinos 1,2-disustituidos. A pesar de esta utilidad este tipo de reacciones son escasas en síntesis de productos naturales. La síntesis del dehidroaltenueno B se realiza mediante una reacción multicomponente de arinos.[32]

Reacciones pericíclicas[editar]
Cicloadiciones [4+2][editar]
Los arinos pueden experimentar reacciones de cicloadición [4+2]. A continuación se muestra el mecanismo concertado de la reacción de Diels-Alder entre bencino y furano. Aun así, posiblemente muchas cicloadiciones [4+2] podrían tener lugar a través un mecanismo por etapas.
Un ejemplo clásico es el de la síntesis del 1,2,3,4-tetrafenilnaftaleno.[33] El tetrabromobenceno puede reaccionar con n-butillitio y furano para formar tetrahidroantraceno.[34] La mezcla des esteroisómeros obtenida se puede separar por su diferente solubilidad en metanol.

Las cicloadiciones [4+2] de arinos se utilizan comúnmente en la síntesis de productos naturales. La limitación principal de esta estrategia es la utilización de dienos con restricciones, tales como el furano o el ciclopentadieno.[3] En 2009 Buszek y colaboradores sintetizaron el herbindole A mediante el uso de una cicloadición [4+2].[35] Así la 6,7-indolina experimenta una cicloadición [4+2] con el ciclopentadieno para obtener un producto tetracíclio complejo.

Cicloadiciones [2+2][editar]
Los arinos experimentan cicloadiciones [2+2] con un amplio rango de olefinas. Debido a la naturaleza electrófila de los arinos, las olefinas con grupos sustituyentes dadores de electrones funcionan mejor en este tipo de reacciones.[36]
Dada la significativa formación de subproductos, estas cicloadiciones son muy poco utilizadas en la síntesis de productos naturales.[3] Aun así algunos existen algunos ejemplos. En 1982 Stevens y colaboradores utilizaron una ciclación [2+2] entre un arino y ceteno acetal para sintetizar el taxodione.[37]

Inserciones en enlace σ[editar]
La reacciones de inserción en enlace σ son un campo propio en el desarrollo de la metodología de arinos. El primer ejemplo de inserción en enlace σ se tiene en la síntesis del compuesto mellein en 1973.[38]

Reacciones catalizadas por metales[editar]
La química de arinos catalizada por metales se encuentra poco desarrollada. Hasta la fecha sólo existe un ejemplo de síntesis total en el que se utiliza una reacción de arinos catalizada por metal.[3] Se trata de la síntesis del producto taiwanin C conseguida por Mori y colaboradores en la que utilizan una catálisis de paladio para realizar un cociclación [2+2+2] entre un arino y un diino.[39]

Otros deshidrobencenos[editar]
Si se denomina el bencino como 1,2-dideshidrobenceno, existen entonces otras 2 posibles isómeros, 1,3-dideshidrobenceno y 1,4-dideshidrobenceno.[40] Sus energías in-silico son respectivamente 106, 122 y 139 kcal/mol (444, 510 y 577 kJ/mol).[41]

La interconversión entre los tres isómeros ha sido estudiada por Maitland.[41][42] La conversión entre las formas 1,2 y 1,3 podría ocurrir con una pirolisis a 900 °C de precursores de fenilarinos. Se requieren temperaturas extremadamente altas para la interconversión de bencinos.

1,4-dideshidrobenceno[editar]
En un expermiento clásico con 1,4-dideshidrobenceno, al calentar a 300 °C, el confórmero [1,6-D2]-A se equilibra rápidamente con el [3,2-D2]-B pero no lo hace con C o con D. La migración simultánea de deuterio a la forma B, y el hecho de que ni C ni D se formen, sólo se puede explicar por la presencia de un intermedio cíclico y simétrico como el 1,4-dideshidrobenceno. Este intermedio se ha identificado en la ciclación de Bergman como etapa determinante de reacción.[43]

La ciclación de Bergman tiene importancia desde el punto de vista biológico desde el descubrimiento de los citostáticos de tipo enediino, estos compuestos tienen la capacidad de escindir las hebras del ADN. Un ejemplo citostático de tipo enediino es la calicheamicina. Ésta se somete a una reacción análoga a la ciclación de Bergman generando un intermedio dirradical que no es otro que el 1,4-dideshidrobenceno. Este dirradical abstrae átomos de hidrógeno de la cadena de ADN y se piensa que esta etapa es fundamental en la actividad biológica de fármacos antitumorales/antibióticos de tipo enediino. Sin embargo las moléculas de tipo enediino son altamente reactivas y poco selectivas, resultando ser poco atractivas como candidatas a fármacos.[44]
Referencias[editar]
- ↑ Gilchrist T.C.; Rees C.W.; (1969) Carbenes, Nitrenes and Arynes Nelson. London.
- ↑ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi 10.1021/cr60216a001
- ↑ a b c d e “Comprehensive History of Arynes in Natural Product Total Synthesis" Tadros & Stoltz, Chem. Rev. 2012, 112, 3550
- ↑ Anslyn, E. V.; Dougherty, D. A.: Modern Physical Organic Chemistry, University Science Books, 2006, p612.
- ↑ a b Gampe, C. M.; Carreira, E. M. Angew Chem. Int. Ed. 2012, 51, 3766
- ↑ Rasziszewski, J. G.; Hess. B. A. J.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52
- ↑ Wenk, H. H.; Winkler, M.; Sander, W. Angew. Chem. Int. Ed. 2003, 42, 502
- ↑ Rondan, N. G.; Domelsmith, L. N.; Houk, K.N. Tetrahedron Lett. 1979, 20, 3273
- ↑ Gilchrist, T. L. Supplement C: The Chemistry of Triple Bonded Functional Groups, Part 1. Patai, S.; Rappaport, Z. Eds., John Wiley & Sons, New York, 1983
- ↑ Hoffmann, R.; Imamura, A.; Hehre, W. J. J. Am. Chem. Soc. 1968, 90, 1499
- ↑ Ueber das 1- und 2-Brom-cumaron. doi:10.1002/cber.19020350286. Consultado el 27 de junio de 2013.
- ↑ Wittig, G., Pieper, G. and Fuhrmann, G. (1940), Über die Bildung von Diphenyl aus Fluorbenzol und Phenyl-lithium (IV. Mitteil. über Austauschreaktionen mit Phenyl-lithium). Berichte der deutschen chemischen Gesellschaft (A and B Series), 73: 1193–1197. doi 10.1002/cber.19400731113
- ↑ Phenyl-lithium, der Schlüssel zu einer neuen Chemie metallorganischer Verbindungen Georg Wittig Naturwissenschaften, 1942, Volume 30, Numbers 46-47, Pages 696-703 doi 10.1007/BF01489519
- ↑ Wittig, G. (1954), Fortschritte auf dem Gebiet der organischen Aniono-Chemie. Angewandte Chemie, 66: 10–17. doi 10.1002/ange.19540660103
- ↑ a b Rearrangement in the reaction of chlorobenzene-1-C14 with potassium amid John D. Roberts, Howard E. Simmons Jr., L. A. Carlsmith, C. Wheaton Vaughan J. Am. Chem. Soc., 1953, 75 (13), pp 3290–3291 doi 10.1021/ja01109a523 10.1021/ja01109a523
- ↑ The Mechanism of Aminations of Halobenzenes John D. Roberts, Dorothy A. Semenow, Howard E. Simmons Jr., L. A. Carlsmith J. Am. Chem. Soc., 1956, 78 (3), pp 601–611 doi 10.1021/ja01584a024
- ↑ Orientation in Aminations of Substituted Halobenzenes John D. Roberts, C. Wheaton Vaughan, L. A. Carlsmith, Dorothy A. Semenow J. Am. Chem. Soc., 1956, 78 (3), pp 611–614 doi 10.1021/ja01584a025
- ↑ Modern Arylation Methods. Edited by Lutz Ackermann 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN 978-3-527-31937-4
- ↑ The Benzyne and Related Intermediates. H. Heaney Chem. Rev., 1962, 62 (2), pp 81–97 doi 10.1021/cr60216a001
- ↑ Wittig, G.; Pohmer, L. Angew. Chem. 1955, 67(13), 348.
- ↑ Radziszewski, J. G.; Hess, Jr. B. A.; Zahradnik, R. J. Am. Chem. Soc. 1992, 114, 52.
- ↑ Wenthold, P. G.; Squires, R. R.; Lineberger, W. C. J. Am. Chem. Soc. 1998, 120, 5279
- ↑ Kukolich, S. G.; Tanjaroon,C.; McCarthy, M. C.; Thaddeus, P. J. Chem. Phys. 2003, 119, 4353
- ↑ Orendt, A. M.; Facelli, J. C.; Radziszewski, J. G.; Horton, W. J.; Grant, D. M.; Michl, J. J. Am. Chem. Soc. 1996, 118, 846
- ↑ a b Warmuth,R. Angew. Chem. Int. Ed. Engl. 1997, 36, 1347
- ↑ Warmuth, R.; Yoon, Acc. Chem. Res. 2001, 34, 96
- ↑ Hoye, T. R.; Baire, B.; Niu, D.; Willoughby, P. H.; Woods, B. P. Nature, 2012, 490, 208
- ↑ a b Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry. University Science Books, 2006.
- ↑ Diemer, V.; Begaut, M.; Leroux, F. R.; Colobert, F. Eur. J. Org. Chem. 2011, 341
- ↑ Kametani, T.; Ogasawara, K. J. J. Chem. Soc., C 1967, 2208
- ↑ Day, J. J.; McFadden, R. M.; Virgil, S. C.; Kolding, H.; Alleva, J. L.; Stoltz, B. M. Angew. Chem. Int. Ed. 2011, 50, 6814.
- ↑ Soorukram, D.; Qu, T.; Barrett, A. G. M. Org. Lett. 2008, 10, 3833
- ↑ Organic Syntheses, Coll. Vol. 5, p.1037 (1973); Vol. 46, p.107 (1966). Link
- ↑ Organic Syntheses, Coll. Vol. 10, p.678; Vol. 75, p.201 Article
- ↑ Buszek, K. R.; Brown, N.; Kuo, D. Org. Lett. 2009, 11, 201
- ↑ Pellissier, H.; Santelli, M. Tetrahedron, 2003, 59, 701
- ↑ Stevens, R. V.; Bisacchi, G. S> J. Org, Chem. 1982, 47, 2396
- ↑ Guyot, M.; Molho, D. Tetrahedron Lett. 1973, 14, 3433
- ↑ Sato, Y.; Tamura,T.; Mori, M. Angew. Chem. Int. Ed. 2004, 43, 2436
- ↑ Wenk, H. H.; Winkler, M.; Sander, W. Angew. Chem. Int. Ed. 2003, 42, 502
- ↑ a b A m-Benzyne to o-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Blake, M. E.; Bartlett, K. L.; Jones, M. Jr. J. Am. Chem. Soc. 2003, 125, 6485. doi 10.1021/ja0213672
- ↑ A p-Benzyne to m-Benzyne Conversion Through a 1,2-Shift of a Phenyl Group. Completion of the Benzyne Cascade, Polishchuk, A. L.; Bartlett, K. L.; Friedman, L. A.; Jones, M. Jr. J. Phys. Org. Chem. 2004, Volume 17, Issue 9 , Pages 798 - 806. doi 10.1002/poc.797
- ↑ Jones, R. R,; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660
- ↑ Bachrach, S. M. Computational Organic Chemistry. John Wiley & Sons, Inc. 2007